
17 April 2021
Integrating Medical Device Product Development, Design for Six Sigma and Quality Systems Requirements — Part 2
Design for Six Sigma (DfSS) is both a business management and a product development process. It uses metrics, data, statistics, team dynamics, risk management and project management tools. Data-driven decision-making processes take products from concept to commercialization. By focusing on design and process parameters, manufacturers can create six-sigma-capable products or services that are based on customer and market needs. In addition—and perhaps more importantly—DfSS enables a company to comply with the medical device quality management system requirements in the product development process.1,2
The six-phase product development process covers the entire product development cycle from idea to eventual sales (see Table I). Notice that each step should end with a risk assessment. Managing risk at each stage helps mitigate and resolve possible problems before they increase costs or present a safety risk for users. Numerous studies have illustrated that having a cross-functional team integrally involved throughout the product development life cycle leads to high success rates in launching sustainable new products.
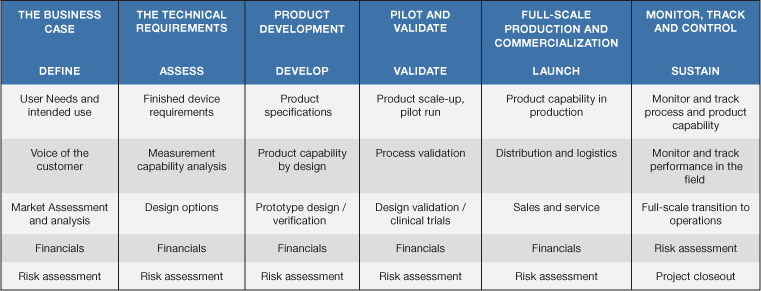
Table II provides an example of how a cross-functional team can be involved in the new product development life cycle. During the process responsibilities shift, and team members need to be able to take on (or give up) levels of responsibility. For example, marketing takes the lead in defining the product development strategy and identifying the applications and customers in the beginning stages of the product development cycle. They are kept informed during the actual design and development of the product but re-emerge in the commercialization stages to drive the marketing initiatives of the new product. To keep the product successful in the market after launch, the marketing department is an active participant.
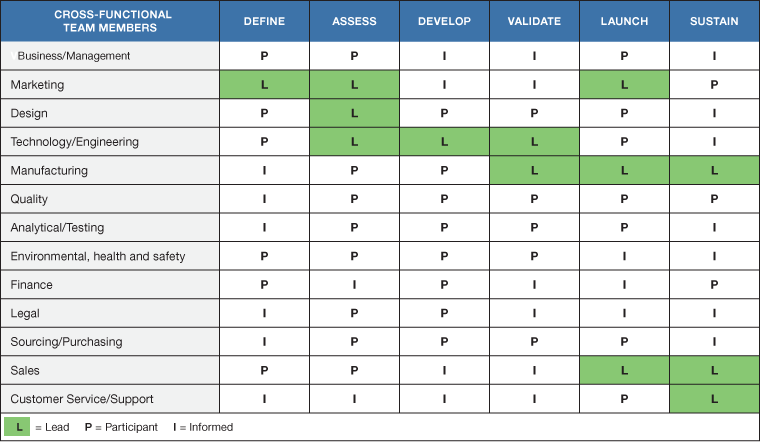
Even though various functions assume the lead in different phases of the product development cycle, the project manager’s role is to maintain continuity. The project manager is the glue that holds it all together.
Medical Device Quality Management System (QMS) Requirements
The medical device Quality Management System (QMS) governs methods used in, and the facilities and controls used for, the design, manufacture, packaging, labeling, storage, installation and servicing of all finished devices intended for human use. It ensures that medical device companies design, develop, manufacture and distribute products that are safe and effective for the patient and the end-user. The requirements include controls related to design, purchasing, production and process, acceptance activities, labeling and packaging, quality and documentation.
Design for Six Sigma can help firms meet the QMS requirements because it incorporates quantifiable metrics, controls, statistical techniques, and documentation. Figure I details a six-stage product development process incorporating the requirements and documentation required for medical devices.
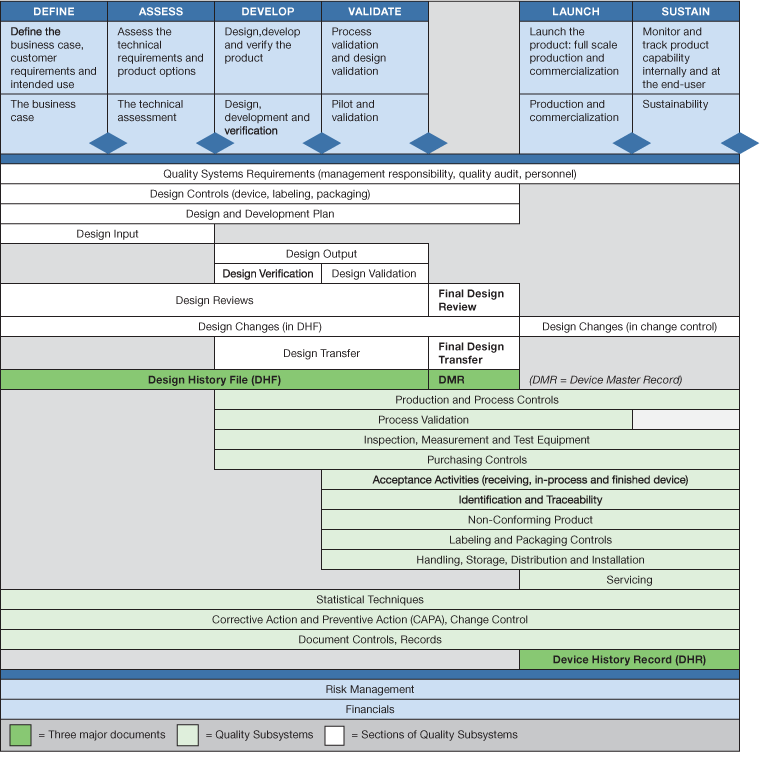
DfSS for QMS
Various Quality Management System elements fit into each phase of a rigorous product development process. These phases can be broken down in a series of steps as follows:
- Define – Define the business case, customer requirements, user needs and intended use of the new product.
- Assess – Determine the technical and product requirements.
- Develop – Design and develop the product and identify the product, component and process specifications. Verify the product meets technical and product requirements.
- Validate – Validate the production process and the validate the device to ensure it meets user needs and intended use.
- Launch – Transfer the product to full-scale production and commercialization.
- Sustain – Monitor performance of product in the field and address any issues when the arise.
Phase 1: Define
The Define Phase is a critical step in crystallizing the fuzzy front end. This involves clarifying goals to get a detailed picture of the market and product and also determining the packaging and labeling requirements for the intended use. One of the biggest challenges for device firms is to identify an unmet need in the market and discern the critical requirements of a device for that intended use. The information is typically analyzed by marketing, with input from design and engineering.
Some of the basic questions that need to be answered for this phase are:
- What unmet need will the new product fulfill for the market, customer or end-user? What are its benefits?
- What are the user needs and usability requirements?
- What is its intended use? How will the end-user use the device? What are the requirements for the intended use (e.g., safety, effectiveness, performance and user interfaces)?
- How does the new product fit into the business strategy? Does this make business sense for the company?
The business case must also include a thorough market and competitive analysis. In Six Sigma terminology, the voice of the customer is used to identify the user needs and intended use, the device performance, and labeling and packaging requirements. Lawrence Friedman gives an excellent treatise on targeting the right markets and aligning with customers in his book Go to Market Strategy 3. Additionally, a detailed analysis of the human factors in both form and function to ensure ergonomic handling, aesthetics, error-free performance and ease of use must not be ignored.4,5
Various methods can be used to obtain the voice of the customer and voice of the market. Methods often include interviews, focus groups, surveys, observation and behavior modeling. Information from the voice of the customer is distilled down to quantifiable finished device performance requirements.
Quality Function Deployment (QFD) takes high level customer needs and correlates those needs with the critical performance specifications of the device. The performance specifications can in turn be correlated to the device design component specifications. The component specifications can then provide the processing controls. Figure II illustrates how the QFD can be used to identify critical production and processing parameters that will produce a consistent product with the desired performance capabilities.
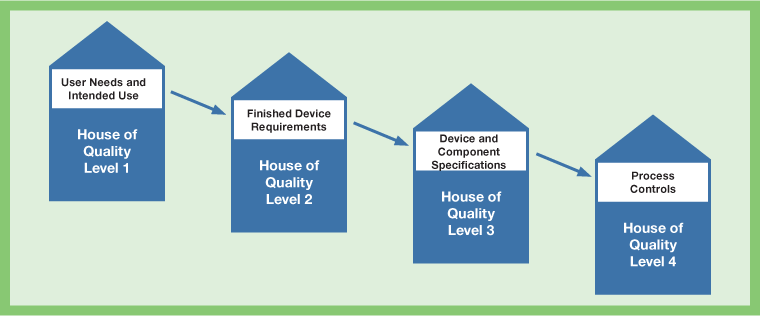
The information from this phase is documented as the design inputs, which are part of the Design History File (DHF).
Phase 2: Assess
The activities of the second phase are focused on identifying technical requirements of the product. Such items can include:
- Detailed product or device requirements and acceptance criteria
- Labeling and packaging requirements
- Sterilization requirements
- Regulatory requirements
- The test measurements for the acceptance criteria, and evaluation of test measurement capability
- Processes for prototyping and design evaluation
- The appropriate development path and plan
Assessment is led by marketing, design and engineering. From the end-user requirements, the product requirements that are critical-to-quality characteristics and tolerances are quantified using the Quality Function Deployment (QFD) technique. Examples include working out the exact dimensions and tolerances, clarity, toughness, electrical outputs, aesthetics and color, and compliance needs of the product. Designing and developing the product becomes much easier once the design inputs are well defined. Design options are whittled down to the one design that meets all the product requirements.
Before developing the product, it is important to ensure that the test measurements used for the acceptance of the components, sub-assemblies and the finished device are capable. Large variations in a measurement system would make it impossible to determine whether or not a product is within specification or out of specification. Standard methods of evaluation are the Gage Repeatability and Reproducibility (GR&R) test or the Precision to Tolerance Ratio (P/T) test. Preference should be given to ANSI, ASTM, ISO or other industry standard test methods.
The outcomes of this phase include criteria for inspection and measurement / test equipment that are part of the Production and Process Control. Product requirements, (i.e., design inputs) are recorded in the DHF. The cross-functional product development team formulates a detailed design and development plan also part of the DHF.
Phase 3: Develop
During the third phase, technology and engineering take the lead in developing the new product based on the device requirements. After a design review, a prototype is developed. The prototype’s specifications, properties and performance (design outputs) are verified with respect to the design inputs.
There are various techniques to evaluate the sigma capability of the product using predictive models and information obtained from statistical techniques. Some of these are design of experiments (DOE), regression analysis, analysis of variance and capability scorecards.
An example of a design capability scorecard is shown in Table III. Model equations are generated based on first principles, modeling or design of experiments. These equations highlight the sensitivities of those components that influence the critical properties of the finished device. The benefit of this type of scorecard is that it identifies those components and parameters that need to be controlled during production. It also assesses the capability of the device based on the existing design. Redesign may be necessary if the prototype is not six sigma capable by design.
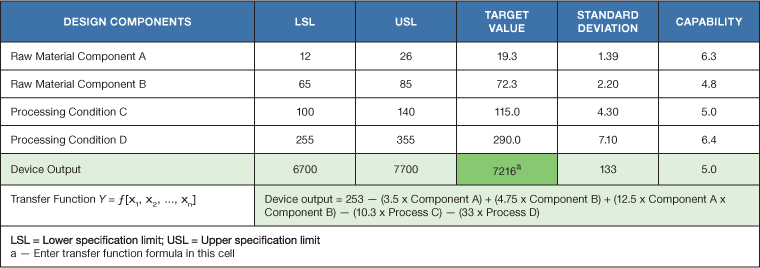
During the product development phase, the hazards, hazardous situations and potential harms resulting from the device design should be identified and evaluated per ISO14971:20196. The risk management document is a living document and must constantly be evaluated and updated if required, throughout the life cycle of the device.
This information is documented for QMS compliance in the DHF and is the source for the design inputs, design outputs, design verification, design validation, design changes and design transfer. The Device Master Record (DMR) is based on the design outputs from design and development.
Phase 4: Validate
In this phase, those processes that cannot be fully verified by inspection and testing need to be validated to ensure the process can consistently meet product specifications. Both process parameters and raw material characteristics must be considered. Evaluation of the finished-device capability is based on critical-to-quality characteristics for the intended use. A capability scorecard details the performance of the influencing components, processing conditions, and device properties. It also provides an assessment of the overall capability of the product (see Table IV). Low-capability components must be identified, and their performance improved via root-cause analysis.
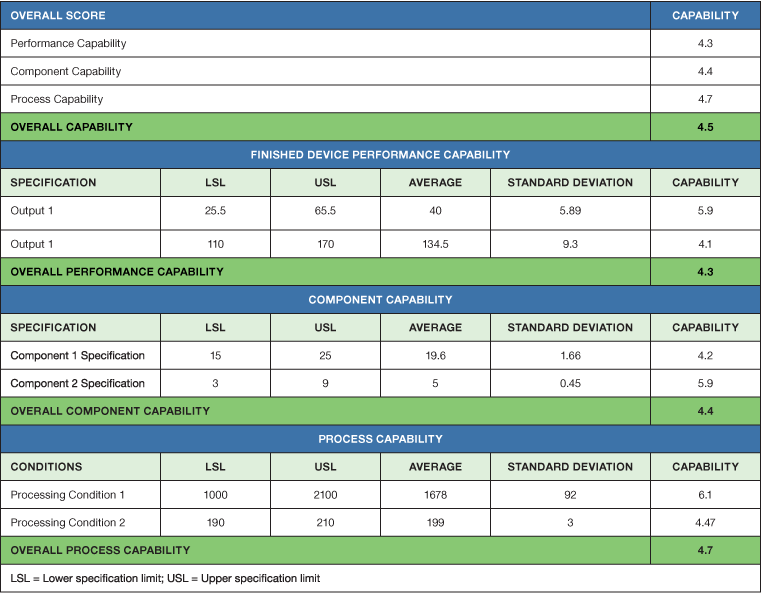
Table IV – Product capability scorecard
The finished device must be evaluated at the end-user or customer to ensure the final product meets the predetermined user needs and intended use (design validation). This stage could take a significant amount of time depending upon the complexity of the design validation at the end-user and any additional clinical trials or evaluation that may be required.
There outputs from the development phase include:
- The DHF includes all the information during the design and development of the device including the design inputs, design outputs, design verification and validation, design reviews, and the design changes.
- The DMR details intended use, specifications for the device outputs and the raw materials, production protocol, packaging and labeling, acceptance activities (raw materials, in-process and finished device), storage, handling, distribution and servicing of the finished device.
Phase 5: Launch
Full-scale production is led by manufacturing and the commercialization process is led by sales and marketing. Product, process and performance capabilities are continually evaluated for six sigma performance in production and at the end-user level.
The intent of Design for Six Sigma is to design the new product to be robust and consistent during production, taking into account the variability of raw materials and process manufacturing. Statistical techniques like statistical process control (SPC), statistical quality control (SQC), and product capability scorecards are used to control and monitor the product and the process.
One of the major factors in a successful product launch is the quality of the product launch itself.7 Giving insufficient resources or attention to marketing and sales often means the product will experience sluggish sales and poor market acceptance. Commercial activities should highlight the intended value of the product.
End-user feedback is monitored to ensure the product meets the user needs and intended use criteria and is safe and effective. For QMS compliance, all production, testing, distribution, installation and servicing data are documented in the Device History Record (DHR) through the life cycle of the product. Changes to processes and product should be captured in the change management and corrective action and preventive action (CA/PA) processes.
Phase 6: Sustain
The finished device should be monitored and tracked through the entire supply chain to evaluate the capability of the entire process. This can be accomplished by setting up a regular audit schedule with a list of critical to quality attributes to monitor. Product capability should be monitored to identify any major shifts and drifts which could result from changes in the process or raw materials over time. The root causes of these shifts and drifts must be identified and corrected. Any changes must be recorded in the change control or CA/PA files.
Product performance in the field should also be monitored, tracked and trended. Any issues identified should be handled via the complaint and adverse event reporting processes. If changes to product or process are required after an investigation, the risk management file of the device must also be reviewed and updated as required.
Before closing out the product development project and making it self-sustainable and operational, the development team should review the results (sales, revenue, quality, margins, product capability, development cycle time etc.) versus the original plan. Thus, information is used in the continuous improvement loop of the quality systems process. The DHR is the repository for all the product data.
Conclusion:
Companies should review their product development processes and assess how to bring rigor and quality regulations compliance into a single business process. Using DfSS tools and methodologies and integrating the QMS within a company’s new product development process can bring structure and clarity, reduced risks, speed-to-market and regulatory compliance. Finished devices can be designed, developed, manufactured, and sold with six sigma capability to meet end-user needs and to maintain their intended use. This leads to product consistency and reliability and sustainable sales, revenues and profits. Simplifying the various development processes into one integrated, manageable process enables better control and compliance.
This may mean changing the way product development is conducted within a company. The challenge is to devise and implement a meaningful, sustainable process that becomes second nature in conducting business and will not be perceived as additional work.
REFERENCES
- 21 CFR 820 Medical Devices – Quality System Regulation – Current Good Manufacturing Practices
- ISO 13485:2016 Medical Devices – Quality Management Systems – Requirements for Regulatory Purposes
- LA Friedman, “Targeting the Right Markets” and “Aligning with Your Customers” in Go to Market Strategy, (New York: Butterworth-Heinemann, 2002)
- IEC 62366-1:2015 Medical Devices – Part 1: Application of Usability Engineering to Medical Devices
- ANSI/AAMI HE75:2009 (R2018) Human Factors Engineering – Design of Medical Devices
- ISO 14971:2019 Medical Devices – Application of Risk Management to Medical Devices
- RG Cooper, Winning at New Products (New York: Basic Books, 2001)