19 September 2021
Focus on the Latest Regulatory and Risk Management Requirements for Medical Plastics
Introduction
The last five years have brought about significant changes to global regulatory and risk management requirements for medical devices, including stringent considerations for the plastics used in these applications. The requirements stem from the European Medical Device and In-Vitro Device Regulations (EU MDR and EU IVDR), the biocompatibility standard ISO 10993:2018 and risk management for medical devices ISO 14971:2019. The General Safety and Performance Requirements in the EU MDR and EU IVDR stipulate several requisites (including risk management) when considering plastics for medical device applications. In addition, biocompatibility must be linked to the risk management process. The FDA has also published a guidance document on biocompatibility and risk management requirements. This post highlights the key regulatory and risk management requirements for plastics and their additives used in medical devices all the way from design and development to processing, manufacturing and use.
After a one-year delay due to the global coronavirus pandemic, the European Union Medical Device Regulation (EU MDR) 2017/745 went into effect on 26 May 2021. The European Union Invitro Diagnostics Regulation (EU IVDR) 2017/746, goes into effect on 26 May 2022. These regulations were introduced to resolve and address a number of deficiencies in the Medical Device Directives (MDD). The EU MDR 2017/745 and the EU IVDR 2017/746 are legally binding regulations across the EU member states. In addition, 1SO14971:2019 was released in December 2019. The following table provides a list of current regulations, standards and guidance documents that specify risk management requirements for medical devices and thus, plastics used in medical device applications.

The Plastic Materials Supply Chain
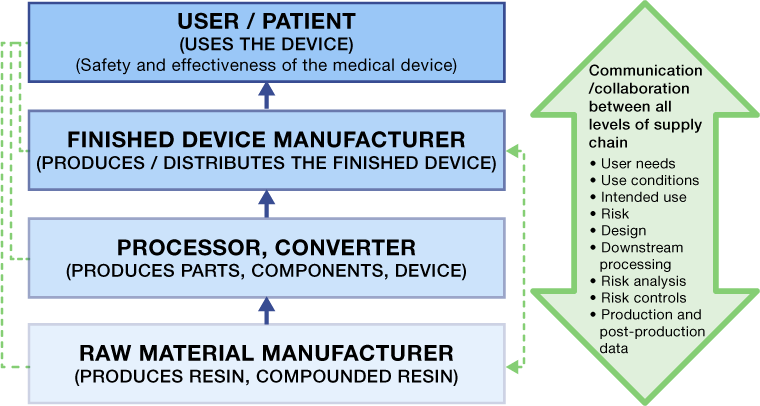
The base plastic resin suppliers can be far removed from the medical device manufacturer. As a result, they may not know the final application and risk associated with their product in the use of the finished device. In addition, they may not be aware of what happens to their resin in terms of additional processing, secondary operations and final assembly. Although the base resin supplier may have provided what they thought was the correct material for the application, these additional processes, heat histories and secondary operations could affect eventual safety and performance of their product during use.
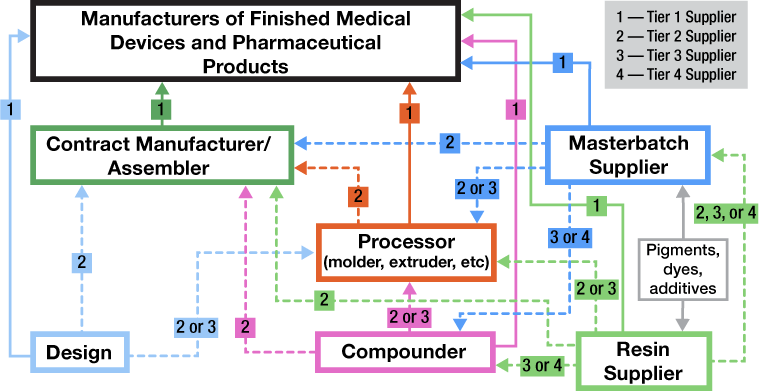
The value chain for plastics suppliers in the medical device industry is illustrated below. The figure details the different levels of where a supplier might fall with respect to how their product is supplied to an OEM. Take, for example, a plastic resin supplier.
Tier 1 supplier: The plastic resin supplier supplies resin directly to the OEM, which molds, assembles, and manufactures all components and the finished device.
Tier 2 supplier: The plastic resin supplier sells resin to a contract manufacturer (a Tier 1 supplier) that molds or makes components or finished devices for the OEM.
Tier 3 supplier: The plastic resin supplier sells resin to a compounder (a Tier 2 supplier) who manufactures and formulates a specialty product, which in turn is sold a contract manufacturer (a Tier 1 supplier), who produces subassemblies or finished devices that is supplied to the OEM.
Tier 4 supplier: The plastic resin supplier sells resin to a compounder (a Tier 3 supplier) who manufactures and formulates a specialty product, which in turn is sold to a molder (a Tier 2 Supplier) who makes a component, which is then sold to a contract manufacturer (a Tier 1 supplier), who produces subassemblies or finished devices that is supplied to the OEM.
[GRAPH]
It is the responsibility of medical device manufacturers to ensure the safety and effectiveness of their finished device. They also need to exert the appropriate levels of controls on their suppliers, depending upon the risk and criticality of the products or services supplied. The extent of control that an OEM can exert on the various levels of the value chain depends on whether the supplier is a direct (Tier 1) or indirect (Tier 2/Tier 3/Tier 4) supplier to the OEM.
EU MDR Requirements with Respect to Materials and Plastics
Annex I of the EU MDR specifies “General Safety and Performance Requirements” versus the older term “Essential Requirements” used in the medical device directive. Annex I, Chapter II details the requirements with respect to design and manufacture of the medical device. The material and plastics considerations during the design and manufacture of the device include:
- Chemical, Physical and Biological properties
- Intended physical and mechanical properties
- The mechanical properties such as strength, ductility, fracture resistance, wear resistance and fatigue resistance
- The impact of production processes (thermal, shear, pressure, radiation etc.) on the intended material properties
- Toxicity and sensitization
- Flammability
- Biocompatibility with biological tissues, cells and body fluids
- Surface properties
- Chemical Resistance to cleaning agents, oils, solvents, disinfectants, lipids etc.
- Potential release of substances or particles, including wear debris, degradation products and processing residues that could affect safety
- The size and the properties of particles which are or can be released into the patient’s or user’s body from the material with specific requirements for nanomaterials
- Justification or rationale for the use of materials that contain carcinogenic, mutagenic or toxic to reproduction (‘CMR’) substances or substances having endocrine-disrupting properties for which there is scientific evidence of probable serious effects to human health
- The rational for the use of phthalates
- Compatibility of materials when in contact with drugs, medicinal products and products of biological origin
- Infection and microbial contamination
- Microbial contamination
- Sterility and methods of sterilization to maintain material and device integrity
- Packaging materials that maintain integrity of the package and sterility and cleanliness of the device
- Interactions with the environment
- Affect and influences of environmental conditions, such as magnetic fields, external electrical and electromagnetic effects, electrostatic discharge, radiation associated with diagnostic or therapeutic procedures, pressure, humidity, temperature, variations in pressure and acceleration or radio signal interferences
- Effect of materials, liquids, and gases, to which the device and materials are exposed during use
- Aging of materials and the effect of safety and performance
- Safe disposal and waste materials
- Protection against radiation
- Compatibility and effect of safety and performance with respect to radiation such as X-Ray, e-beam and ionizing radiation
- Active devices and devices connected to them
- Effect of electromagnetic interference
- Electrical leakage and electrical shocks / insulating properties
- Active implantable devices
- Compatibility and effects on the use of energy sources with particular reference, where electricity is used, to insulation, leakage currents and overheating of the devices
- Excessive increase of leakage currents
- Degradation and aging of the materials used
- Excess heat generated by the device
- Protection against mechanical and thermal risks
- Mechanical risks connected with, for example, resistance to movement, instability and moving parts
- Effect of vibrations on safety and performance
- Effect of electricity, gas or hydraulic and pneumatic energy supplies
- Moving parts durability, wear and frictional properties
- Equipment housing requirements
Risk Management Requirements and Process
Risk Management for medical devices is focused on the use of the device and the risks associated with that device to the user and/or patient. Therefore, the hazards and risks during normal use, must be identified, evaluated, and removed or reduced to acceptable safety levels. Risk Management focuses on:
- Normal use conditions,
- The use environment, and
- Users
This is important because the supplier of a plastic resin may be far removed from the production of a finished device, and may not know whether or not their product could potentially contribute to a hazardous situation leading to patient harm. The risk management process includes the following steps:
- Identification of hazards associated with the use of the device
- Determining the sequence of events that could lead to a hazardous situation (all the way from design, raw materials, production, packaging, sterilization, storage, distribution and use)
- Evaluating the risk and harms to patient or user from the hazard and the resulting hazardous situation
- Removing, reducing, mitigating and/or managing the risk via design, material, production and other relevant risk control measures
- Determining that the residual risk and overall residual risk is acceptable
- Monitoring the effectiveness of the risk controls
Potential Hazards Associated with Plastics
Potential hazards associated with plastic materials are related to the ability or inability of the material to reduce or protect user and patient from:
Energy hazards like:
- Acoustic energy (infrasound, ultrasonic)
- Electric Energy (electric fields, current leakage, magnetic fields, static discharge, voltage)
- Mechanical energy (heavy/falling objects, moving parts, vibrating parts)
- Potential / stored energy (bending, kinking, compression, cutting, shearing, suspended mass, tension, torsion)
- Radiation energy (gamma, X-Ray, infrared, laser, microwave, ultraviolet)
- Thermal energy (high temperature, low temperature)
Biological and chemical hazards like:
- Biological agents (bacteria, fungi, parasites, prions, toxins, viruses)
- Chemical agents (acidic, alkaline, oxidants, flammable, combustible, solvents, cleaning agents, heavy metals, particles)
- Biocompatibility (toxicity, carcinogenicity, allergenic, immunosuppressive, irritants, sensitizing)
Environmental hazards like:
- Particulates
- Temperature
- Humidity
Usability hazards like:
- Slips (using soft-touch, flexible materials)
- Use of incorrect device type or size (by using color coding)
- Use of excessive force (by using material that have lubricity)
- Misguiding of devices during procedures (by using radiopaque materials)
Sequence of Events Leading to Hazardous Situations During Use
Hazards and the sequence of events leading to hazardous situations can occur anywhere from the design, through production, packaging, sterilization, storage, distribution and to use as shown in the figure below. This s also true of biological hazards. No matter where the hazard originated, the user or patient is exposed to the hazardous situation that could lead to harm including death or serious injury.
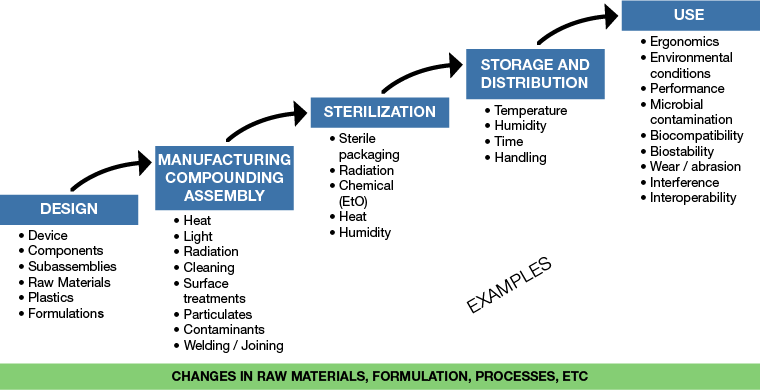
As an example, particulates in a device that could lead to patient injury or harm during the device’s use could originate from:
- The raw material resin itself
- During production
- During packaging
- During use with the abrasion of material with another product
Depending upon where the source of the particulate came from, appropriate controls need to be implemented. These controls could be on the material cleanliness itself, controls in production processes or the production environment, controls in material packaging or the packaging environment or the use of non-abrading devices or procedures.
Effective Communication Between the Supply Chain
In order to design and manufacture safe and effective devices, medical device OEMs must communicate effectively across the entire plastics supply chain to ensure all suppliers know and understand what they need to do to ensure that their specific products or processes do not expose the user and patient to any hazardous situations.