7 June 2021
European Union Medical Device Regulation (EU MDR) and Risk Management — Reduce, Eliminate, Minimize, Remove or Avoid Risk?
After a one-year delay due to the global coronavirus pandemic, the European Union Medical Device Regulation (EU MDR) 2017/745 went into effect on 26 May 2021. The regulation was introduced to resolve and address a number of deficiencies in the two Medical Device Directives (MDD) — the Active Implantable Medical Device (AIMD) Directive — 90/385/EEC established in 1990 and the Medical Device Directive (MDD) — 93/42/EEC established in 1993. The EU MDR 2017/745 consolidates both these directives into one medical device regulation. The EU MDR 2017/745 is a legally binding regulation across the EU member states.
The regulation has a major focus on safety and risk management, post-market surveillance activities, and specific requirements for notified bodies. Article 10, “General Obligations to Manufacturers”, requires manufacturers to ensure compliance to their Quality Management Systems and establish, implement, document and maintain a system for risk management as described in Section 3 of Annex I.
Requirements for Risk Management in the Regulation
Sections 1 to 5 of Annex I (General Safety and Performance Requirements), Chapter I (General Requirements) clearly layout the requirements for risk management. These include:
- Ensure that devices during normal use are suitable for their intended use
- Risks which may be associated with device use constitute acceptable risks when weighed against the benefits to the patient
- Reduce risk as far as possible. The requirement to reduce risk as far as possible means reduction of risks without adversely affecting the benefit-risk ratio
- Establish, implement, document and maintain a risk management system that includes
- A risk management plan
- Identifying and analyzing known and foreseeable hazards
- Estimating and evaluating risks during normal use and foreseeable misuse
- Eliminating or controlling risks
- Evaluating the impact of information from the production phase and the post-market surveillance system with respect to hazards, the frequency of occurrence, estimates of their associated risks and on the overall risk, benefit-risk ratio and risk acceptability. Amend risk control measures if required.
- Employ risk control measures with the priority of:
- Safe design and manufacture
- Adequate protection measures
- Information for safety
- Eliminate or reduce risk related to use error
- Minimize all known and foreseeable risks and undesirable risks to acceptable levels when weighed against the evaluated benefits to the patient and/or user
- Design for patient safety
- Ensure that risk control measures conform to safety principles, taking into account general state of the art.
- Reduce and manage risks so that the residual risk associated with each hazard as well as overall residual risk is judged acceptable.
- Ensure risk is evaluated during design and manufacture, per Annex I, Chapter II — General Performance and Safety Requirements
Key elements with respect to risk management in the EU MDR regulation include:
- Normal use conditions and foreseeable misuse
- Usability and use error
- Reducing risk as far as possible
- Risk acceptability
- Residual risk for each hazard
- Overall residual risk
- Benefit-risk analysis
- Post-market activities
Regulation EU MDR 2017/745 and its relationship to ISO 14971
The risk management requirements in Annex I, Chapter l of the regulation mirror those detailed in ISO 14971. Although the regulation does not specifically mention the medical device risk management standard ISO 14971, it does require compliance to harmonized standards. Recital 22 states “compliance with harmonized standards as defined in Regulation (EU) No 1025/2012 of the European Parliament and of the Council (2) should be a means for manufacturers to demonstrate conformity with the general safety and performance requirements and other legal requirements, such as those relating to quality and risk management, laid down in this Regulation.” Article 2 (70) defines a harmonized standard as “a European standard as defined in point (1)(c) of Article 2 of Regulation (EU) No 1025/2012”. The document Commission Implementing Decision (EU) 2020/437 of 24 March 2020 on the harmonized standards for medical devices drafted in support of Council Directive 93/42/EEC published the Official Journal of the European Union lists all the standards applicable to medical devices. This includes EN/ISO 14971, and also includes other relevant standards like EN/IEC 62366, EN/ISO 10993, EN/IEC 60601 and EN/IEC 62304.
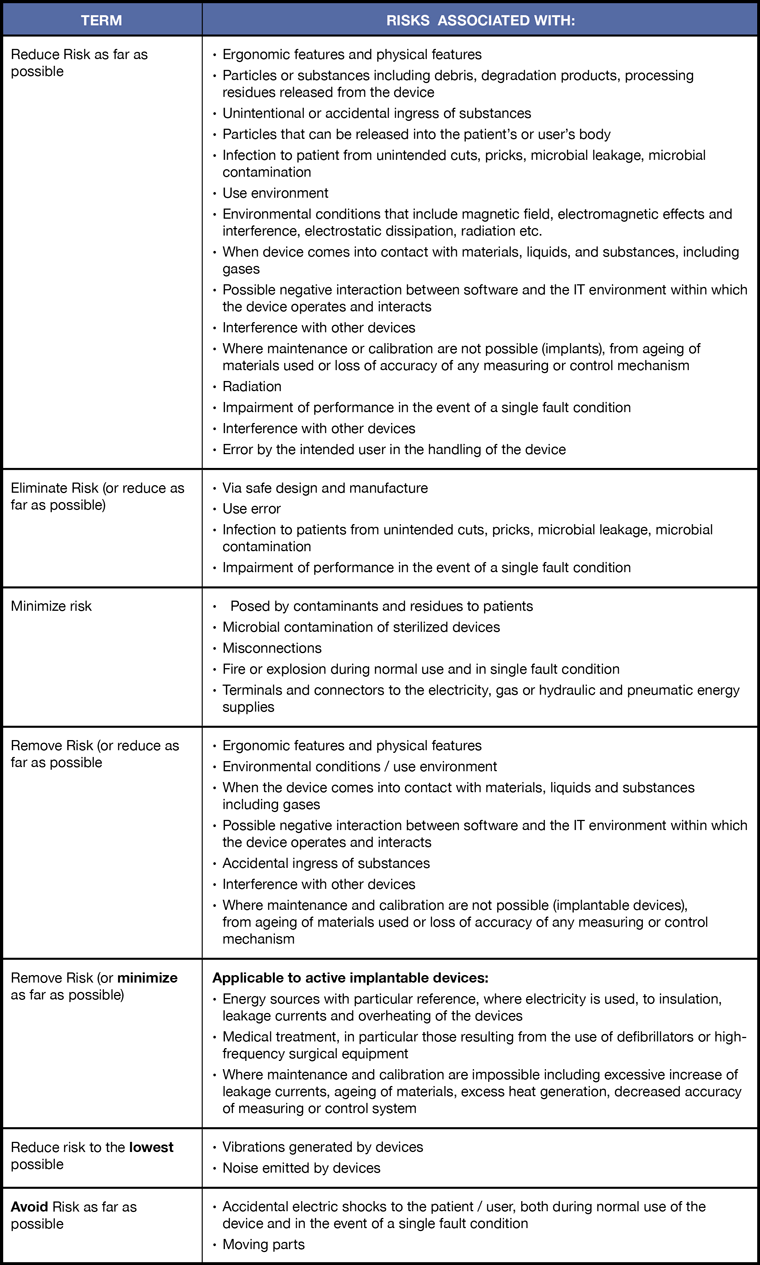
Reduce, Eliminate, Minimize, Remove, Avoid Risk?
One of the more confusing issues with respect to risk management in the regulation are the different requirements with respect to reducing risk. The details for each term and corresponding requirements are summarized in the table. Some of the terms used are:
- Reduce risk as far as possible
- Eliminate risk (or reduce as far as possible)
- Minimize risk
- Remove risk (or reduce as far as possible)
- Remove risk (or minimize as far as possible)
- Reduce risk to the lowest possible
- Avoid risk to the lowest possible
How does a manufacturer attempt to address all these different requirements for risk?
In Clause 4.2 of ISO 14971:2019, Note 1 states that “The manufacturer’s policy for establishing criteria for risk acceptability can define the approaches to risk control: reducing risk as low as reasonably practicable, reducing risk as low as reasonably achievable, or reducing risk as far as possible without adversely affecting the benefit-risk ratio.”
Therefore, it is up to the manufacturer to determine the processes by which they are going to manage risks based on all the different requirements listed above and in the table. The manufacturer should clearly establish policies and processes describing how their methods of reducing risk will comply with all the different requirements listed in the regulation.
In summary
The EU MDR regulation has a very strong emphasis on risk management. Risk is mentioned 243 times in the regulation when compared to a combined total of only 69 times in the two medical device directives — Active Implantable Medical Device 90/385/EEC (14 times) the Medical Device Directive 93/42/EEC (55 times). Although ISO 14971 is not cited in the regulation, it is a documented harmonized standard in the Official Journal of the European Union. The regulation requires compliance with harmonized standards. Requirements and processes for risk management in the regulation mirror those detailed in ISO 14971:2019. Several (at times confusing) terms are used in the regulation to address risk. The manufacturer must clearly define — in policies and procedures — how they will address these various methods of reducing, eliminating, minimizing, removing and avoiding risk.
Regulation EU MDR 2017/245 — The different terms used for risk and the corresponding design and manufacture elements associated with those terms